Chemistry of Complex Materials
Chemically active metal oxide surfaces and interfaces ‒such as catalysts, sensors and electrodes‒ play a crucial role in our society and in the development of new technologies. In fact, more than 80 % of industrial processes worldwide, and all the electro-chemical energy sources, rely on the chemical surface activity of (nanostructured) catalysts to function. In most cases, the mechanisms are unknown, preventing controlled improvement of these processes. Here computational condensed-matter chemistry can be of immense benefit in the exploration of the nanoworld and its possibilities to improve our world.
The picture below illustrates the eSSENCE project Chemistry of Complex Materials. We recently developed efficient reactive force-fields to enable large-scale simulations of metal-oxide nanoparticles (2015, upper left picture). Experimentalists have discovered that aggregation of octahedral nanoparticles of this material readily occurs (upper right figure). With the new force-field we, too, find such agglomeration, which allows us to discover exiting new features such as their lifetimes, stability and reactivity (bottom two figures which are snapshots from our MD simulation).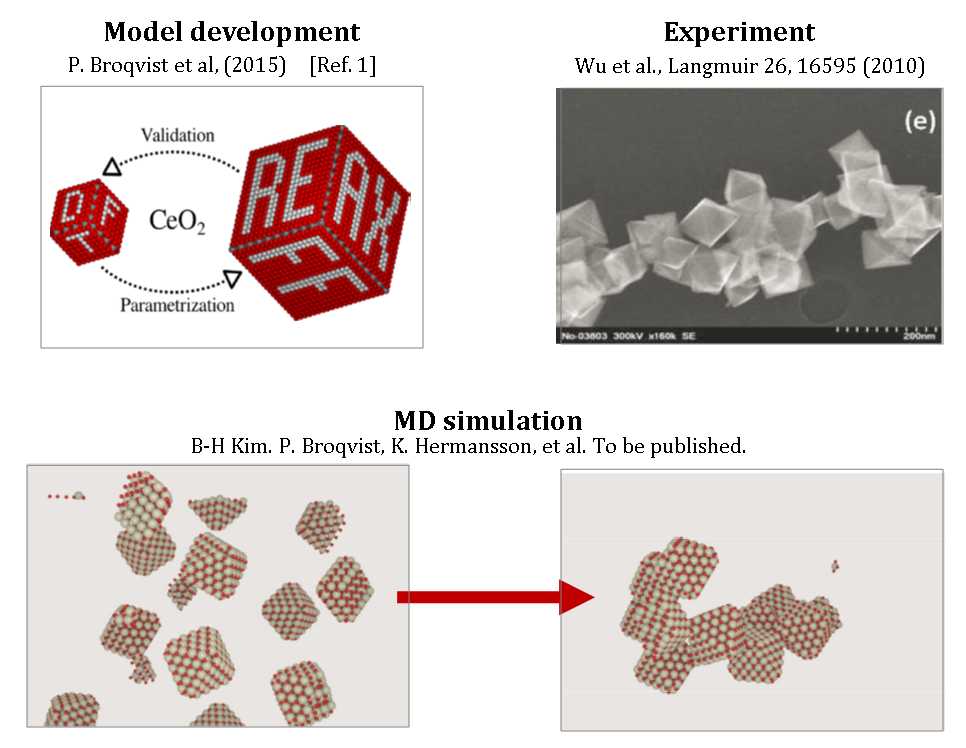
Aims
Realistic modelling of chemically active materials, and their surfaces and interfaces is challenging because:
- Large-scale computational models are needed to accommodate the complexity of realistic, non-perfect surfaces and nano-particles.
- In this field, the computational scientist needs to make shrewd decisions concerning both the structural model for the surface/interface and the total-energy method to use. The former aspect is an additional complication compared to, say, calculations for molecules or perfect crystals.
- As we here deal with chemical phenomena and red-ox reactions, the electronic structure can be very complicated to model accurately and needs special care.
- Temperature/entropic effects are often crucial to incorporate to achieve realistic results.
The aim of this project is dual: to make progress in the method and model development for simulations concerning the chemistry of complex materials, and to use these methods to derive unique information about such systems and processes.
Methods
We are heavily involved in the development of multiscale modelling methods for complex materials. We combine a range of theoretical methods, including wave-function based QM calculations, DFT of various flavours (e.g. [2],[3]), tight-binding-DFT (of the so-called SCC-DFTB type), and reactive force-field models [1]. The SCC-DFTB method, for example, is about two orders of magnitude less computationally expensive compared to a standard DFT calculation. The accuracy and transferability of SCC-DFTB models crucially depend on a set of parameters, which have to be optimized. An efficient scheme for this was presented in Ref. 4.
Our multi-scale modelling research forms the thrust of our methodological efforts towards making “calculations meet reality”. But we also wish to make “calculations meet experiments”. Adequate realistic models for post-processing of simulation data is as important as the data generation itself, since it links directly to spectra, images, etc. produced from the experiments. If this is successfully achieved, the calculations offer added insight that goes beyond what experiments can give. Our simulated IRRAS spectra of the CO/TiO2(110) system compared to experiment is a good example [5].
Research group
PI:Prof. Kersti Hermansson
Department of Chemistry – Ångström, Uppsala University
Dr. Peter Broqvist
Department of Chemistry – Ångström, Uppsala University
Dr. Pavlin Mitev
Department of Chemistry – Ångström, Uppsala University
Dr. Jolla Kullgren
Department of Chemistry – Ångström, Uppsala University
Links and references
- P. Broqvist, J. Kullgren, M. J. Wolf, A. C. T. van Duin, K. Hermansson, “A ReaxFF force-field for ceria bulk, surfaces and nanoparticles”, J. Phys. Chem. C 119, 13598 (2015)
- I. Beinik, M. Hellström, Th. N. Jensen, P. Broqvist,J.V. Lauritsen, NATURE COMMUNICATIONS, Article no. 6:8845, DOI: 10.1038/ncomms984 (2015)
- G. G. Kebede, D. Spångberg, P. D. Mitev, P. Broqvist, K. Hermansson, J. Chem. Phys., in press (2016).
- M. Hellström, K. Jorner, M. Bryngelsson, S.E. Huber, J. Kullgren, Th. Frauenheim, P. Broqvist, J. Phys. Chem. C, 2013, 117, 17004 (2013).
- S. Hu, Z. Wang, A. Mattsson, L. Österlund, K. Hermansson, J. Phys. Chem. C 119, 5403 (2015).