A multiscale modelling platform for Materials Chemistry
On request from the European commission, the newly created European Materials Modellling Council (EMMC) conducted a survey in the spring of 2016, aimed at identifying what are the most important obstacles hindering a wider use of materials e-science in European industry. It was found that the largest bottleneck hindering a wider use of modelling today is the lack of appropriate models to treat systems of large (realistic) complexity. Models of high accuracy and speed were requested as well as techniques to couple and link models and data over time and length scales (multiscale modelling).
Within the eSSENCE framework we are building a “simulation environment” to automatize the parameterization processes involved in multiscale modelling. The picture below illustrates this effort.
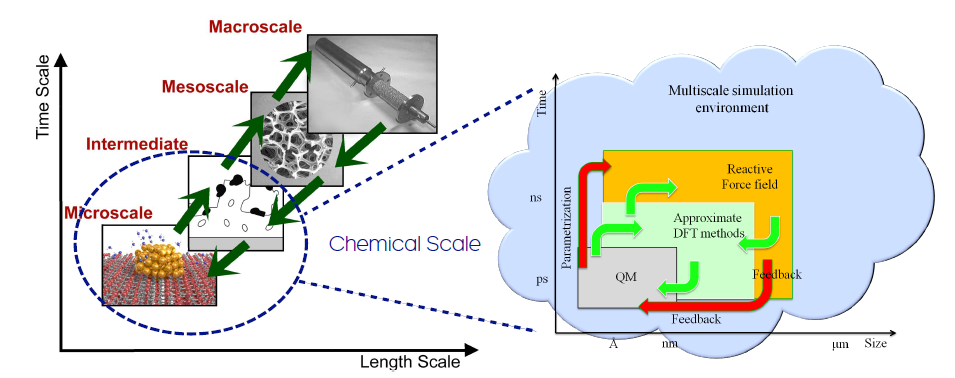
Aims
The aim of this project is to provide a computational tool to move efficiently along the multiscale ladder, while at the same time losing as little chemical information as possible in the process.
Methods
This simulation Environment will provide links between selected theoretical methods, and an interactive interface is being developed, which utilizes the versatile Atomistic Simulation Environment (ASE), which is a suite of Python programming modules and libraries for the creation and visualization of atomistic models and for handling atomistic simulations. So far, we are focussing on the tight-binding and reactive force-field codes and the parametrizations that shall connect them seamlessly. We combine a range of theoretical methods, including wave-function based QM calculations, DFT of various flavours, tight-binding-DFT (of the so-called SCC-DFTB type), and reactive force-field models. The SCC-DFTB method, for example, is about two orders of magnitude less computationally expensive compared to a standard DFT calculation. But, the accuracy and transferability of SCC-DFTB models crucially depend on a set of parameters, which have to be optimized with some skill. In this effort we are therefore collaborating with experts in numerical algorithms, namely
- Professor Per Lötstedt at Department of Information Technology at Uppsala University and
- Dr. Eddie Wadbro at the Department of Computing Science at Umeå Universityas well as with main code developers abroad, namely
- the DFTB+ program group at the Bremen Center for Computational Materials Science, headed by Professor Thomas Frauenheim (tight-binding methods), and
- the ReaxFF group headed by A. C. T. van Duin at Penn State University in the US (reactive force-fields).
Research group
PI:Prof. Kersti Hermansson
Dept. of Chemistry-Ångström, Uppsala University
Project leader: Dr. Peter Broqvist
Dept. of Chemistry-Ångström, Uppsala University
eSSENCE collaborators:
Prof. Per Lötstedt
Scientific Computing, Uppsala University
Dr. Eddie Wadbro
Computing Science, Umeå University