Non-adiabatic chemical processes: chemistry beyond the Born–Oppenheimer approximation
Non-adiabatic chemical reactions have many novel applications. For instance the chemiluminescence of luciferin is used extensively in medical research, as a versatile and non-invasive probe tool to monitor a variety of processes. Further examples of non-adiabatic chemistry are found in fields like photosynthesis and solar cells. Understanding and modeling these reactions is crucial for gaining the ability to control and design new materials, molecular systems and catalysts.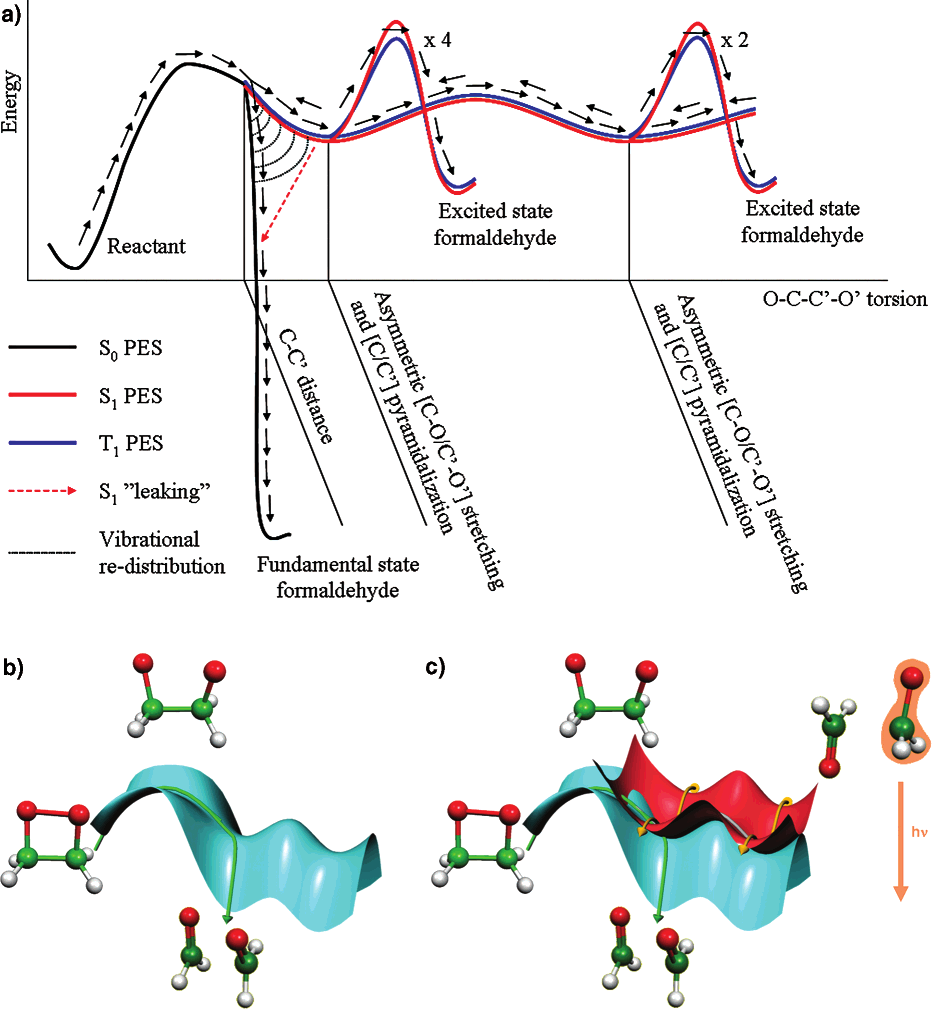
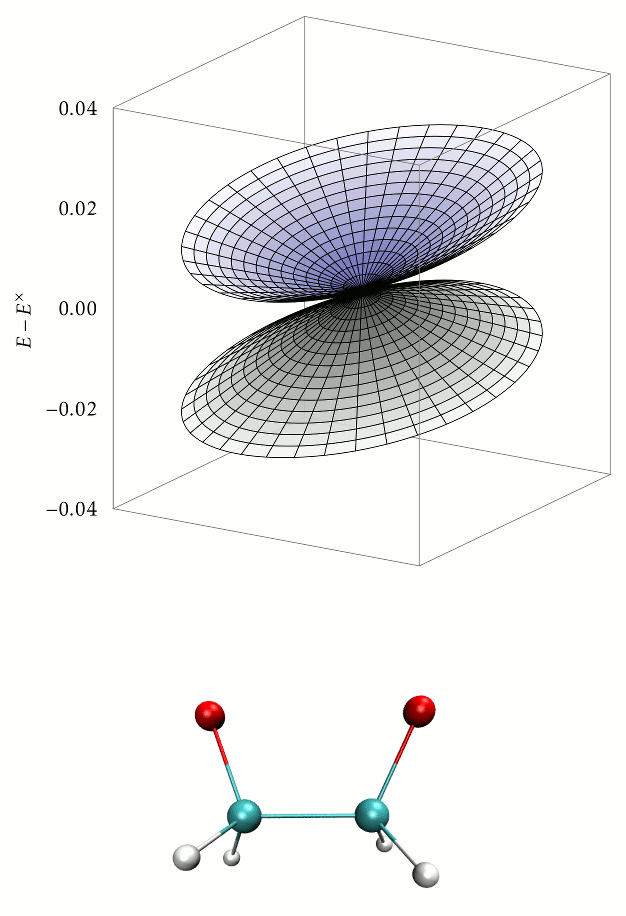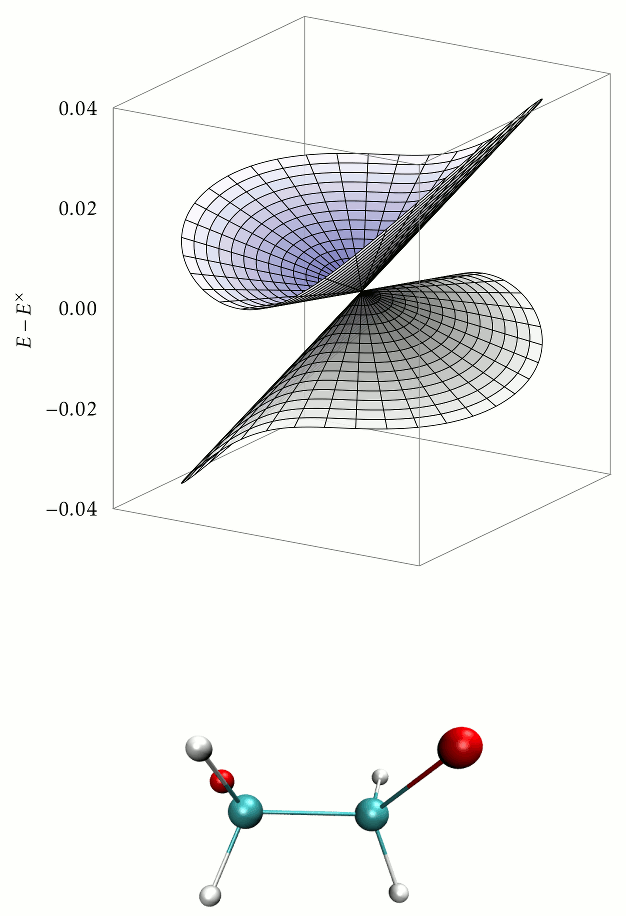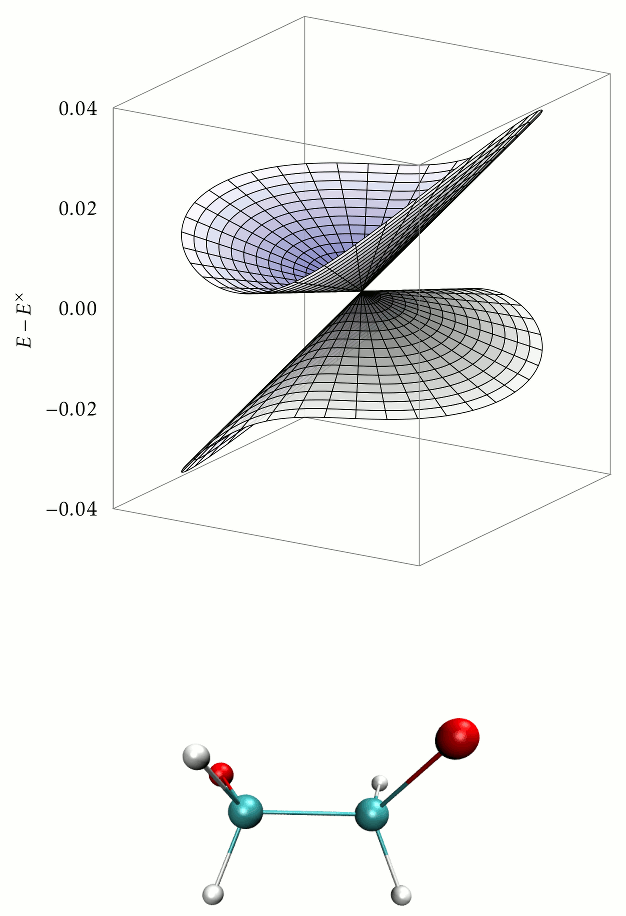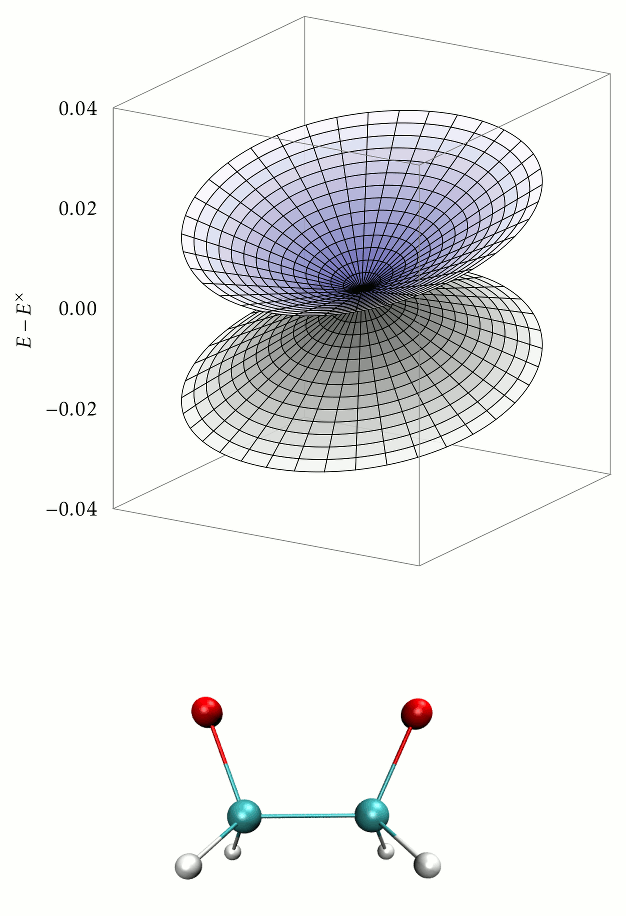
Aims
Our purpose is understanding the details of non-adiabatic chemical processes, with particular focus on chemiluminescence and bioluminescence. We want to be able to characterize these systems and explain and their behavior from a chemical point of view. In order to rationalize their properties, differences and trends, we also develop the necessary theoretical and computational methods, tools and protocols.
Methods
Thanks to the “digital revolution” of the last decades, theoretical and computational chemistry has become an estabished and respected field of science with predictive capabilities, and an essential tool for interpreting the physical world. We use the methods derived from quantum chemistry to unveil the details of non-adiabatic reactions. In particular, to obtain a wavefunction description for the electronic degrees of freedom we use multiconfigurational methods and perturbation theory. The dynamics of the reaction are treated through classical or semi-classical simulations. Recent developments in multiconfigurational methods and computational techniques allow us to perform more accurate and realistic simulations than one could think of a few years ago.
Research group
PI:Prof. Roland Lindh
Department of Chemistry – Ångström, Uppsala University
Ignacio Fernández Galván
Department of Chemistry – Ångström, Uppsala University
Links and references
The Chemistry of Bioluminescence: An Analysis of Chemical Functionalities. I. Navizet, Y.-J. Liu, N. Ferré, D. Roca-Sanjuán, R. Lindh, Chem. Phys. Chem. 12 (2011) 3064-3076. DOI: 10.1002/cphc.201100504
A surface hopping algorithm for nonadiabatic minimum energy path calculations. I. Schapiro, D. Roca-Sanjuán, Roland Lindh, Massimo Olivucci, J. Comput. Chem. 36 (2015) 312-320. DOI: 10.1002/jcc.23805