Monte Carlo approach to proteins in cellular environments
Biophysical characterizations of proteins are typically conducted under dilute conditions. However, in cells, proteins are exposed to a thick “soup” of macromolecules. An important question, therefore, is how macromolecular crowding affects the properties of proteins, such as their thermal stability. Recent experiments indicate that these effects cannot be understood in terms of volume exclusion alone. For a fundamental understanding of the effects caused by macromolecular crowding, there is a need for novel computational approaches.
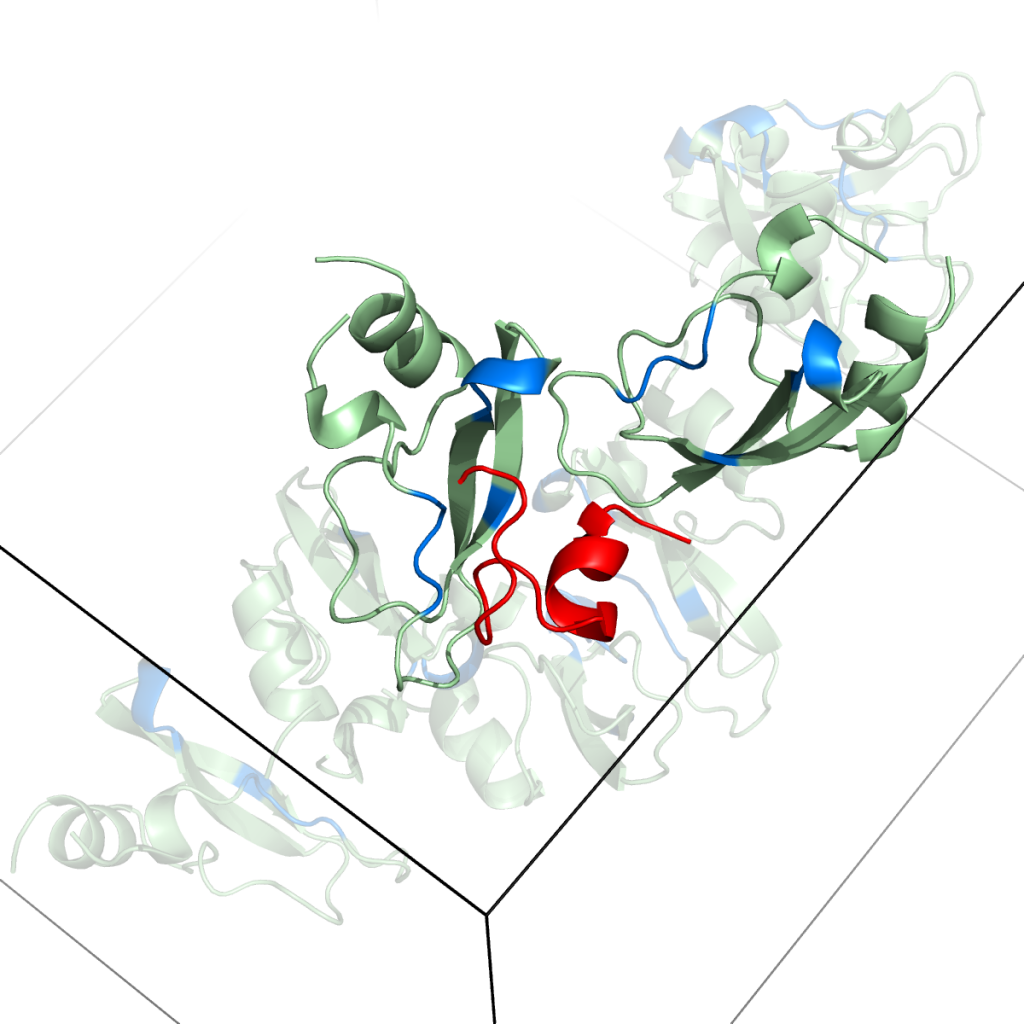
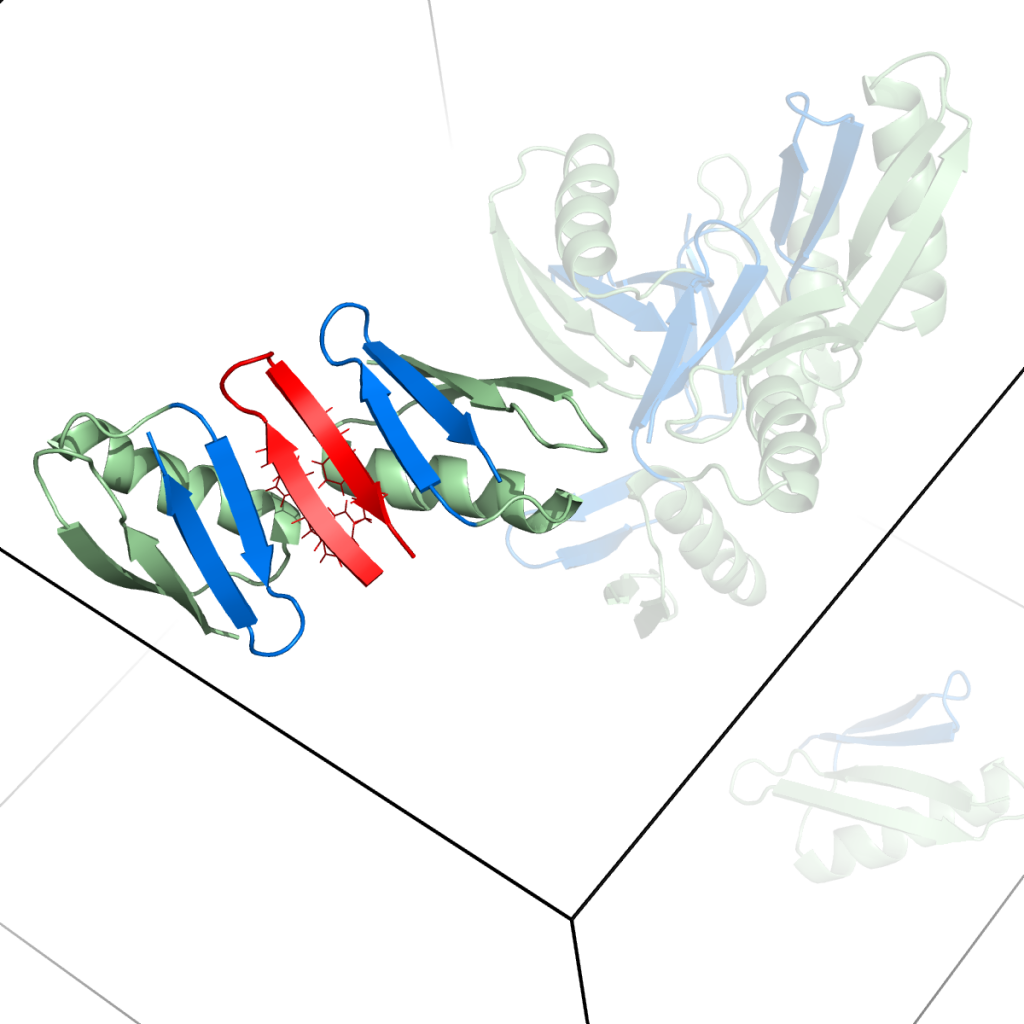 Snapshots from simulations of peptides with protein crowders [1].
Snapshots from simulations of peptides with protein crowders [1].
Aims
In this project, we will develop techniques for simulating proteins in the presence of interacting macromolecular crowders. The methods will be used to investigate the effects of macromolecular crowding on biophysical processes such as protein folding, binding and oligomerization, and to elucidate the nature of the interactions causing these effects. Knowledge of these effects is essential in order to understand how proteins operate in a cell.
Methods
Our computational approach is based on Monte Carlo techniques and an all-atom protein model along with an implicit solvent force field. This model has previously been used to study the folding of many peptides and small proteins under dilute conditions. To this model, we will add crowder molecules in a computationally tractable, yet detailed representation. With this model and state-of-the-art simulation techniques, we aim to create a platform for simulations with interacting macromolecular crowders. A first test of the approach was recently reported [1].
Research group
PI:Prof. Anders Irbäck
Department of Astronomy and Theoretical Physics, Lund University
Links and references
J. Chem. Phys. 143, 175102 (2015)
2. A. Bille, S. Mohanty and A. Irbäck
J. Chem. Phys. 144, 175105 (2016)